

The Spike Protein and Systemic Prion Disease: RBD Prion-Like Domain/Mitochondrial Interactions Induce a Novel Form
From infection to gene therapy, the Spike Protein fills organs with non-functioning aggregates.


Hypothetical mechanistic illustration of prion and SARS-CoV-2 mitochondrial targeting. Prions are infectious proteinaceous particles that have been functionally associated with the progression of major human neurodegenerative disorders such as Creutzfeldt-Jakob disease. Empirically elucidated modes of actions of infectious prions include multiple inhibitory patterns adversely affecting normative neural communication as well as initiation of apoptotic or necrotic neural damage. Furthermore, a likely mechanism of prion-related neural dysfunction may also involve aberrant restitution of native conformation of abnormally folded cellular proteins. These pathophysiological effects may be potentially due to loss of normative proteasome functioning due to enhanced reactive oxygen species associated with compromised mitochondria energy metabolism. Infectious prion diseases may also induce memory, personality and movement abnormalities as well as depression, confusion and disorientation. Interestingly, some of these same behavioral sequelae have also been observed to occur subsequent to COVID-19 and may also share pathogenic mitochondrial damage caused by SARS-CoV-2 infection with subsequent production of ROS or RNS. Taken together, we speculate that pathophysiological effects of long COVID may involve the induction of spontaneous production of infectious prion species. Interestingly, mitochondria may represent the central focus of both induced disorders
A case report was published on July 2nd of this year which confirmed a long-standing hypothesis of mine: SARS-CoV-2 and its Spike Protein induce prion disease. The case involves an elderly gentleman who experienced a delayed onset of Creutzfeldt-Jakob disease (CJD) over a year post COVID infection. The timeline is greatly disturbing.
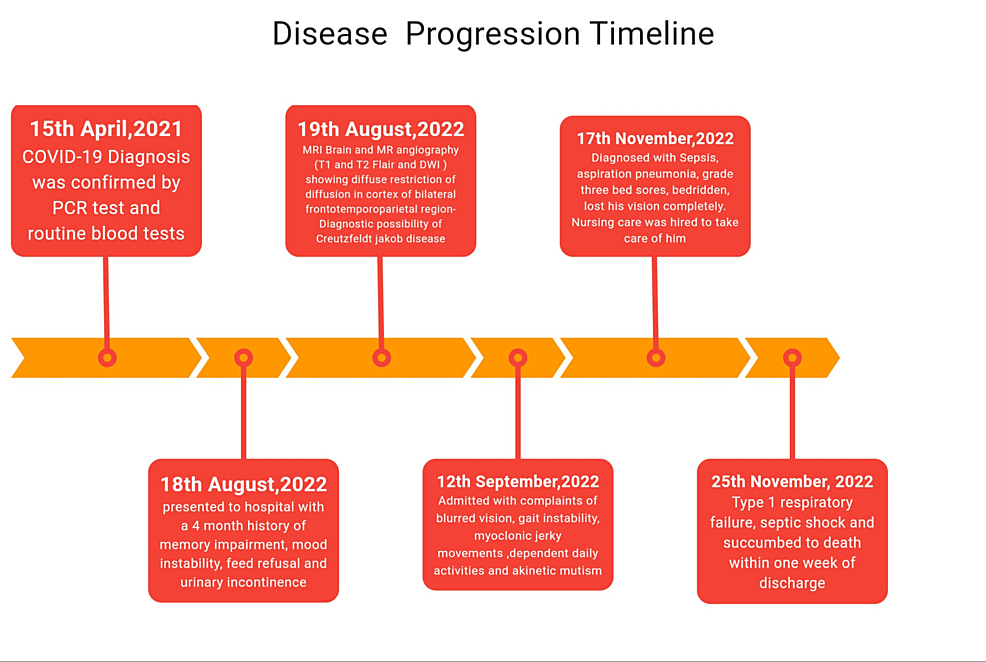
Unusually Late Onset of Creutzfeldt-Jakob Disease Following COVID-19 Infection in India: A Case Report
https://www.cureus.com/articles/263323-unusually-late-onset-of-creutzfeldt-jakob-disease-following-covid-19-infection-in-india-a-case-report#!/
However, after much additional research and contemplation, it finally dawned on me. We are not dealing with just CJD – the “traditional” prion disease. No. I propose we are dealing with a novel form of prion disease which is not limited to the traditional neurodegenerative course. To understand the concept, imagine a cellular game of tag. Proteins you touch become “it.” The identical proteins they touch become “it.” And they can self-replicate. How? The RBD of the Spike Protein is the catalyst for this phenomenon. It contains a prion domain. Additionally, as the initial graphic explains, the Spike Protein’s inflammatory properties can also induce misfolding and prion disease.
A comprehensive study using bioinformatics has identified a large number of viral proteins from diverse species that have prion-like signatures in their genetic sequence. In particular, they identified prion-like domains in viral surface proteins involved in receptor binding and fusion with the host cell [19]. These same authors later published a paper analyzing the spike protein's prion-like potential. They found a prion-like domain in the RBD of the SARS-COV-2 spike protein, which was missing from the original SARS-CoV virus. Asparagine (N) and glutamine (Q)-rich regions are characteristic features of many prion proteins.
And, more importantly:
A study evaluating the amyloidogenic potential of the spike protein used both theoretical and experimental methods to verify that the SARS-CoV-2 spike protein can cause amyloid-like fibrils to appear after the protein has been subjected to proteolysis. Theoretical predictions identified seven potentially amyloidogenic sequences within the spike protein. In laboratory experiments where the protein was incubated with the protease neutrophil elastase, amyloid-like fibrils appeared during 24 hours of coincubation. A specific segment, spike 194-213 (FKNIDGYFKI), was highly abundant after six hours, and it overlapped almost completely with the most amyloidogenic sequence identified theoretically. Neutrophils responding to immune activation release neutrophil elastase into the medium, where it would have access to the spike protein and be able to break it down into the amyloidogenic segments [29]. The SARS-CoV-2 spike protein has a unique polybasic four-amino-acid insert, RRAR, at the junction of the S1 and S2 segments, which is not present in SARS-CoV. This sequence has several cleavage sites susceptible to proteases such as neutrophil elastase [30]. Neutrophil elastases secreted by neutrophils attracted to the sites of inflammation caused by the SARS-CoV-2 spike protein can presumably cleave the encountered spike protein and release the S1 segment into the circulation, potentiating an amylogenic response.
The authors were bold enough to state what we already almost certainly knew regarding the vaccines. Why was this not headline news?
We have examined the extensive literature concerning the prion-like properties of the SARS-CoV-2 spike glycoprotein. Furthermore, we identify pathways through which the mRNA vaccines could be capable of delivering the spike protein to the brain, which we suggest happens via exosomes released from germinal centers in the spleen traveling up the vagus nerve, increasing the risk of neurodegenerative disease.
A Potential Role of the Spike Protein in Neurodegenerative Diseases: A Narrative Review
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9922164/
Prions are also thought to be a factor in Long COVID. Again, the authors of this study are focusing on the traditional neurodegenerative aspect of prion disease.
In this regard, we hypothesize that illnesses that have been attributed to the syndrome known as long-term COVID-19 may actually originate, in part, from spontaneous prion production. We note that while fully developed prion disorders are universally fatal, misfolded proteins that accumulate in response to SARS-CoV-2 infection can most likely be cleared after a brief delay. By contrast, in the absence of another immunological challenge, the clearance mechanisms are severely overwhelmed in the setting of full-blown prion disease. Put simply, virus infection alters the process via which prions reproduce. Although SARS-CoV-2 has not yet been widely localized in the CNS, its’ damage has been associated with the infection (Douaud et al. 2022; Stein et al. 2022). This phenomenon warrants additional attention given the clinical similarities exhibited by long-term COVID-19 and prion diseases.
Potential Prion Involvement in Long COVID-19 Neuropathology, Including Behavior
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10047479/
This is where I propose that we need to look far beyond the traditional neurodegenerative disease of the prion. We need to look at it in a new light. That this process may apply to ALL organs. I will now provide some examples from current research.
HEART DISEASE SEQUELAE
Case reports have described prion associated cardiomyopathy and case control studies support the premise that heart disease is both a cause and consequence of prion disease. These clinical observations are supported by our preliminary observations in mouse models of prion disease.
Project 1. Should we worry about prion associated heart disease?
https://biomedicalsciences.unimelb.edu.au/sbs-research-groups/microbiology-and-immunology-research-groups/lawson-lab-neurodegenerative-caused-by-prions/project-1.-should-we-worry-about-prion-associated-heart-disease
DIABETES SEQUELAE
Type 2 diabetes (T2D) is a highly prevalent metabolic disease characterized by chronic insulin resistance and β-cell dysfunction and loss, leading to impaired insulin release and hyperglycemia. Although the mechanism responsible for β-cell dysfunction and death is not completely understood, recent findings suggest that the accumulation of misfolded aggregates of the islet amyloid polypeptide (IAPP) in the islets of Langerhans may play an important role in pancreatic damage. Misfolding and aggregation of diverse proteins and their accumulation as amyloid in different organs is the hallmark feature in a group of chronic, degenerative diseases termed protein misfolding disorders (PMDs). PMDs include highly prevalent human illnesses such as Alzheimer’s and Parkinson’s disease, as well as more than 25 rarer disorders. Among them, prion diseases are unique because the pathology can be transmitted by a proteinaceous infectious agent, termed a prion, which induces disease by propagating protein misfolding and aggregation. This phenomenon has a striking resemblance to the process of protein misfolding and aggregation in all of the PMDs, suggesting that misfolded aggregates have an intrinsic potential to be transmissible. Indeed, recent studies have shown that the pathological hallmarks of various PMDs can be induced in vivo under experimental conditions by inoculating tissue extracts containing protein aggregates into animal models. In this review, we describe our current understanding of the molecular mechanism underlying the prion-like transmission of protein aggregates and its possible role in T2D
Prion-Like Protein Aggregates and Type 2 Diabetes
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5411686/
THE CLOTS
Interestingly, PrP was found to accelerate fibrin-rich clot formation, which was resistant to plasmin-mediated fibrinolysis, consistent with enhanced thrombus stability provoked by PrP.
Fibrinogen Mitigates Prion-Mediated Platelet Activation and Neuronal Cell Toxicity
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8977893/
I believe you now fully understand my concerns. We are almost certainly looking at an entirely new form of prion disease. One that is not limited to the traditional neurodegenerative disease. This new version attacks multiple proteins, multiple organs.
We need biopsies and autopsies to determine to what extent, if any, this is occurring. My fear is that there is a silent wave of prion disease building which will catch (some of) us by surprise.
Thank you for your support, dialogue and readership. I will research therapeutics that may mitigate this phenomenon.
I may have figured out why the spike protein can cause an aggressive form of CJD. The receptor binding domain of SARS-CoV-2 spike has an amino acid sequence, YQAGS, that closely resembles YQRGS in the C-terminal domain of the prion protein. Antibodies to YQAGS are very effective at protecting from disease, but they could bind to the prion protein through molecular mimicry. It has been shown that antibodies to the C-terminal domain of the prion protein cause an aggressive form of CJD that is not due to prion protein misfolding but rather to its clearance. This also suggests that it is a LOSS of FUNCTION that is more important than a GAIN of FUNCTION, in the process of neurodegeneration in CJD. We talk about this idea, with references, in the Cureus paper at this link.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9922164/.
Thank you for this post Walter and for your ongoing relentless search for understanding of just what we are really dealing with here. I absolutely echo your concern, it would certainly appear we are seeing a new level of prion disease that goes far beyond what we thought we knew vis a vis CJD and similar conditions. We knew that the Spike was prionogenic, but this is all several orders of magnitude worse and may explain to a significant degree what we are observing in many who are experiencing multiple organ/system damage & dysfunction. May God help us all.