

SARS-CoV-2 Manipulates All Six Signaling Pathways of Oncogenic Viruses
The Spike Protein alone manipulates four of them and may manipulate all, as well.

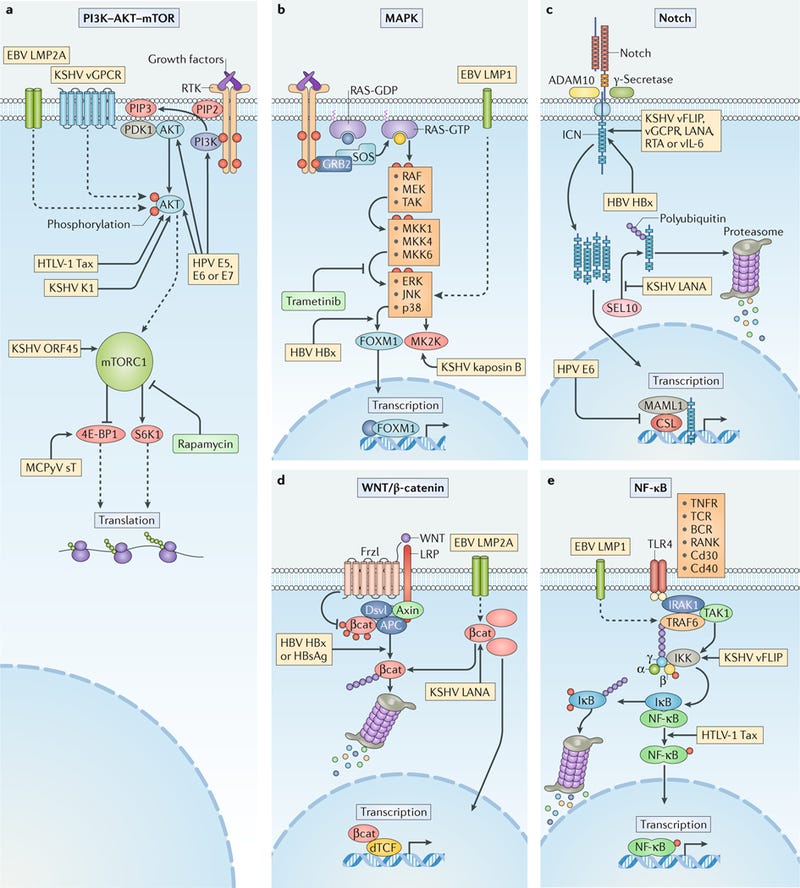
Human oncogenic viruses modulate signal transduction pathways that control cell growth, proliferation and survival to optimize cellular conditions for viral replication, virion assembly and autophagic evasion in the absence of growth or survival signals. Dysregulation of these pathways through mutation or viral factors has been implicated in many cancers. Targeting of critical axes in these pathways by human oncogenic viral factors is indicated by yellow boxes. Arrows represent activation, whereas blocking arrows represent inhibition. Dashed arrows indicate activation or promotion with multiple steps not shown. a | Mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) is a master regulator that coordinates biomolecule availability and stress stimuli to yield tuned responses that promote cell growth and inhibit autophagy. Growth factor binding to receptor tyrosine kinases (RTKs) regulates mTORC1 activity through phosphatidylinositol 3-kinase (PI3K) and the serine/threonine kinase AKT. Ligand-bound RTKs autophosphorylate and recruit PI3K to the plasma membrane, where it converts phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 recruits 3-phosphoinositide-dependent protein kinase 1 (PDK1) and AKT. Multiple viruses modulate the activity of the AKT pathway and downstream components, such as eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1) and ribosomal protein S6 kinase β1 (S6K1). b | The mitogen-activated-protein kinase (MAPK) pathway is also activated by ligand-bound RTKs. Autophosphorylated tyrosine residues bind SH2 domains of growth factor receptor-bound protein 2 (GRB2), which localizes the guanine-exchange factor son-of-sevenless (SOS) to the inner membrane. SOS allows for the exchange of GDP for GTP on RAS. Activated GTP-bound RAS initiates a MAPK cascade, which activates transcription factors such as forkhead box protein M1 (FOXM1) and additional effectors such as MK2 kinase (MK2K). Together, they enhance the expression of pro-survival and pro-inflammatory genes through increased transcription and stabilization of mRNAs, respectively. c | A conformational change in Notch when bound to ligands on neighbouring cells enables sequential cleavages by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and γ-secretase. Cleavage releases intracellular domain of Notch (ICN) into the cytoplasm, where it can translocate to the nucleus and coordinate the transcription of proliferation and differentiation-related genes with DNA-bound CSL protein and the co-activator mastermind-like 1 (MAML1). ICN is downregulated by SEL10 polyubiquitylation-mediated proteasomal degradation. d | β-Catenin (βcat) is inactivated in a complex with adenomatous polyposis coli gene product (APC) and axin, which phosphorylates βcat and targets it for proteasomal degradation. Upon WNT glycolipoprotein binding to extracellular domains of prolow-density lipoprotein receptor related protein 1 (LRP1) and frizzled (Frzl), dishevelled (Dsvl) is recruited to the cytoplasmic domain of Frzl. Subsequent phosphorylation of LRP sequesters axin and prevents degradation of βcat. Accumulating βcat translocates to the nucleus, where it co-activates Drosophila T cell factor (dTCF)-mediated transcription of cell growth genes. e | Several immunity-related cell surface receptors, including Toll-like receptor 4 (TLR4) and tumour necrosis factor receptor (TNFR), activate the canonical nuclear factor-κB (NF-κB) pathway when bound to their respective ligands. TLR4 activation leads to phosphorylation and recruitment of interleukin-1 receptor-associated kinase 1 (IRAK1) to the adaptor protein myeloid differentiation primary response protein MYD88. A complex containing the E3-ubiquitin kinase TNF receptor-associated factor 6 (TRAF6) forms, which generates a scaffold for the polyubiquitin-binding NF-κB essential modulator (NEMO) of inhibitors of NF-κB (IκB) kinase (IKK). Orphan nuclear receptor TAK1 (also known as NR2C2) activates IKK, which then phosphorylates the inhibitory subunit (IκB) and targets it for polyubiquitylation and proteasomal degradation. A conformational change between the NF-κB subunits p50 and p65 allows activating phosphorylation and translocation to the nucleus, where it induces expression of inflammatory and pro-survival genes. BCR, B cell receptor; E5, E6, E7, early proteins 5, 6 and 7; EBV, Epstein–Barr virus; ERK1, extracellular-signal-regulated kinase 1; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HBx, HBVX protein; HPV, human papilloma virus; HTLV-1, human T-lymphotropic virus 1; JNK, JUN N-terminal kinase; KSHV, Kaposi sarcoma-associated virus; LANA, latency-associated nuclear antigen; LMP, latent membrane protein; MCPyV, Merkel cell polyomavirus; MEK, MAPK/ERK kinase; MKK, mitogen-activated protein kinase kinase; RAF, RAF proto-oncogene serine/threonine-protein kinase; RANK, receptor activator of NF-κB (also known as TNFRSF11A); RTA, replication and transcription activator; sT, small tumour antigen; Tax, transactivator from X-gene region; TCR, T cell receptor; vFLIP, viral FLICE inhibitory protein; vGPCR, viral G protein-coupled receptor.
All the world stands aghast at the sudden dramatic increase in cancers. This new surge often involves particularly aggressive cancers termed “turbo cancers.” For reasons which can only be intuited as nefarious, the medical establishment and MSM fail to address the most obvious and almost certain cause of these cancers: SARS-CoV-2 and its Spike Protein.
The above image is from a paper published in 2018. It discusses in detail the molecular mechanisms by which viruses promote the initiation and development of cancer. I will now demonstrate how SARS-CoV-2 manipulates each of the five signaling pathways implicated above, and how it also manipulates the DNA damage response. In essence, we have virus which appears to be custom-tailored to induce cancer in its victims.
Viral infection is a major contributor to the global cancer burden. Recent advances have revealed that seven known oncogenic viruses promote tumorigenesis through shared host cell targets and pathways. A comprehensive understanding of the principles of viral oncogenesis may enable the identification of unknown infectious aetiologies of cancer and the development of therapeutic or preventive strategies for virus-associated cancers. In this Review, we discuss the molecular mechanisms of viral oncogenesis in humans. We highlight recent advances in understanding how viral manipulation of host cellular signalling, DNA damage responses, immunity and microRNA targets promotes the initiation and development of cancer.
Molecular mechanisms of viral oncogenesis in humans
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6336458/
In early 2022, I published an article where I hypothesized that the Spike Protein of SARS-CoV-2 acts like an oncogenic “flash drive,” complete with “instructions” to induce and/or accelerate existing cancers.
By looking at the Spike Protein’s interactome, we can see a landscape where it would seem inevitable that aggressive cancer would be initiated or an existing, perhaps even occult cancer, become hyper aggressive.
AN ONCOGENIC “FLASH DRIVE”: THE SPIKE PROTEIN OF SARS-CoV-2 AND CANCER
https://wmcresearch.org/an-oncogenic-flash-drive-the-spike-protein-of-sars-cov-2-and-cancer/
Now that we have the evidence, let’s examine each of the five signaling pathways mentioned above and how SARS-CoV-2 and its Spike Protein manipulates them in an oncogenic manner.
Pi3K–AKT–mTor signalling pathway
Through western blotting and RNA analysis, we found increased mTOR signaling and suppression of genes related to autophagy, lysosome, and vesicle fusion in Vero E6 cells infected with SARS-CoV-2 as well as in transcriptomic data mining of bronchoalveolar epithelial cells from severe COVID-19 patients.
Increased mTOR Signaling and Impaired Autophagic Flux Are Hallmarks of SARS-CoV-2 Infection
https://www.mdpi.com/1467-3045/45/1/23
MAPK signalling pathway
NF-κB signalling pathway
SARS-CoV-2 spike S1 protein and live SARS-CoV-2 virus have each been shown to activate MEK/MAPK signaling in lung vascular cells [35,43]. SARS-CoV-2 infection of A549 cells with live SARS-CoV-2 virus in a recent study resulted in activation of MAPK ERK1/2 signaling [35]. Those findings are expanded in our data in Figure 2a, with the novel finding that a MEK1/2 MAPK ERK1/2 inhibitor blocks the S1 spike-stimulated production of IL-1β in A549+ lung epithelial cells [43]. In addition, the same study showed that SARS-CoV-2 virus infection of A549 cells stimulates NF-kB p65 activation [35]. Other recent studies have shown that S1 spike can activate NF-kB p65 in A549 cells [49]. Therefore, we next sought to determine whether the S1 spike protein alone was sufficient to stimulate MAPK ERK1/2 (p-ERK1/2) or NF-kB (p-p65) activation in our A549+ lung cells, using western blotting of A549+ cell lysates to measure phosphor-ERK1/2 MAPK (activated) as well as NF-kB phosphor-p65 (activated).
The SARS-CoV-2 S1 Spike Protein Promotes MAPK and NF-kB Activation in Human Lung Cells and Inflammatory Cytokine Production in Human Lung and Intestinal Epithelial Cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9607240/
Notch signalling pathway
Here, we describe and examine predictions of a model in which NOTCH may represent a central signaling axis in lung infection in Coronavirus Disease 2019 (COVID-19). A pathway involving NOTCH signaling, furin, ADAM17, and ACE2 may be capable of increasing SARS-CoV-2 viral entry and infection. NOTCH signaling can also upregulate IL-6 and pro-inflammatory mediators induced to hyperactivation in COVID-19. Furthermore, if NOTCH signaling fails to turn down properly and stays elevated, airway regeneration during lung healing can be inhibited—a process that may be at play in COVID-19.
NOTCH signaling in COVID-19: a central hub controlling genes, proteins, and cells that mediate SARS-CoV-2 entry, the inflammatory response, and lung regeneration
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9386183/
WNT/β-catenin signalling pathway
Cytokine profiles observed in COVID-19 patients have revealed increased levels of IL-1β, IL-2, IL-6, and TNF-α and increased NF-κB pathway activity. Recent evidence has shown that the upregulation of the WNT/β-catenin pathway is associated with inflammation, resulting in a cytokine storm in ARDS (acute respire distress syndrome) and especially in COVID-19 patients. Several studies have shown that the WNT/β-catenin pathway interacts with PPARγ in an opposing interplay in numerous diseases. Furthermore, recent studies have highlighted the interesting role of PPARγ agonists as modulators of inflammatory and immunomodulatory drugs through the targeting of the cytokine storm in COVID-19 patients. SARS-CoV2 infection presents a decrease in the angiotensin-converting enzyme 2 (ACE2) associated with the upregulation of the WNT/β-catenin pathway.
Interplay of Opposing Effects of the WNT/β-Catenin Pathway and PPARγ and Implications for SARS-CoV2 Treatment
https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2021.666693/full
DNA damage response
Here, by using an in vitro cell line, we report that the SARS–CoV–2 spike protein significantly inhibits DNA damage repair, which is required for effective V(D)J recombination in adaptive immunity. Mechanistically, we found that the spike protein localizes in the nucleus and inhibits DNA damage repair by impeding key DNA repair protein BRCA1 and 53BP1 recruitment to the damage site.
SARS–CoV–2 Spike Impairs DNA Damage Repair and Inhibits V(D)J Recombination In Vitro
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8538446/
The above paper was “retracted.” This was done against the vehement wishes of the authors to keep the paper in good standing. I have studied the paper extensively and can find no reason why it should have been retracted.
Given the overwhelming amount of evidence presented (and there are far more papers indicating similar findings) I find it beyond absurd that the horse is unable to find its cart. We need to focus on attenuating these pathways in the presence of Spike and SARS-CoV-2. Needless to say, it is a monumental task. Especially when you consider the plethora of other pathologies induced by the Spike and SARS-CoV-2.
I will continue to search for further understanding and solutions.
Thank you. Your support, readership and dialogue all make my work possible.
Fascinating. So are we also to conclude that the mRNA "vaccines" which turn our bodies into a spike protein factory, are also "custom-tailored to induce cancer"? In which case, shouldn't be seeking to bring those responsible for this devastating two-pronged attack on humanity to justice?
Thank you for your research and dedication to revealing the dark corners of our government research. When Nixon started a “war on cancer”, we now know what he meant, and poured billions of tax payers money into. When Reagan started a “war on drugs”, we now know how government got funding for its black ops. When Clinton started “free trade” it transferred 7 million blue collar jobs to China, and created the northern rust belt, opioid epidemic, and lives of despair. Where would we all be without government for the he people, of the people?