

Reviewing “Persistent Vascular Complications in Long COVID: The Role of ACE2 Deactivation, Microclots, and Uniform Fibrosis”
Published last week, the most convincing evidence to date supporting my SPED (Spike Protein Endothelial Disease) hypothesis.

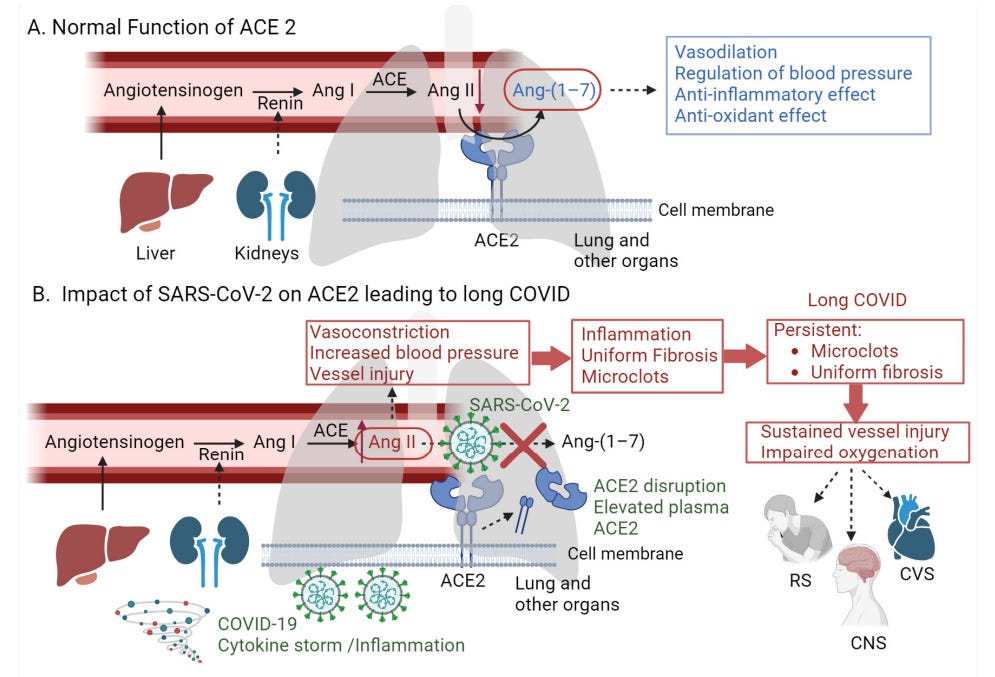
A graphical representation of the proposed hypothesis. (A) Normal function of ACE2 in the renin–angiotensin system (RAS): Renin converts Angiotensinogen to angiotensin I (Ang I). Ang Infect. Dis. Rep. 2024, 16 565 I is then converted to angiotensin II (Ang II) by ACE, which is present on the surfaces of endothelial cells, primarily in the lungs and kidneys. ACE2 acts as a counter-regulator by converting Ang II to angiotensin 1–7 (Ang-(1–7)), which exhibits vasodilatory and anti-inflammatory activities, thus regulating blood pressure. (B) Impact of SARS-CoV-2 on ACE2 leading to long COVID: SARS-CoV-2 binds to ACE2, reducing its presence on the endothelium of the lung and other organs, which disrupts the normal function of ACE2. This increases Ang II levels, leading to vasoconstriction, vessel injury, and inflammation. Moreover, the binding of SARS-CoV-2 to ACE2 causes the shedding of these receptors by various proteases, leading to elevated levels of plasma ACE2. These events contribute to the formation of persistent microclots and uniform fibrosis, leading to impaired oxygenation—a hallmark symptom of long COVID that affects various systems, including the respiratory (RS), central nervous (CNS), and cardiovascular (CVS) systems (created with BioRender.com, accessed on 1 April 2024).
First, I would like to start with the most significant statement in this paper, published last week, which offers the most convincing evidence for my SPED hypothesis:
Similarly, mice infected with the SARS-CoV virus, or its S protein, exhibited severe symptoms like those of ACE2-deficient mice, including worse outcomes from acid-induced lung damage characterized by structural changes in the lung tissue, increased lung edema, and enhanced leukocyte presence.
I have said this from the beginning. All of my research indicated to me that the Spike Protein itself would induce the endothelial dysfunction found in SARS-CoV-2 infections, long COVID and eventually vaccine injury.
Three years ago, I published the following:
SARS-CoV-2 IS PRIMARILY A DISEASE OF THE SPIKE PROTEIN’S INTERACTION WITH THE MICROVASCULATURE
https://wmcresearch.org/the-spike-protein-and-the-microvasculature/
This is what I would end of calling Spike Protein Endothelial Disease (SPED). I maintain it is the basis for slow, progressive long term organ destruction which the evidence shows is occurring in those with Long COVID. Tests need to be conducted to determine in what percentage of the population this may occur.
Part of SPED, I hypothesized, involves the induction of microclots, which would form aggregates (amyloid properties) and be resistant to degradation. The paper we are reviewing shows that this is, indeed, the case.
Importantly, introducing purified, recombinant SARS-CoV-2 S1 spike protein into normal plasma capable of clotting can trigger the formation of abnormal clots. These clots not only assume amyloid states but also show resistance to fibrinolysis, the process that breaks down blood clots.
Another important aspect of SPED is the loss of ACE2. I had also predicted that the Spike Protein would induce this phenomenon, again, three years ago.
COVID-19 IS A DISEASE OF OVEREXPRESSED ANG II: ACE2 DEGRADING/DOWNREGULATION BY THE SPIKE PROTEIN RESULTS IN PRIMARY ARTERIAL HYPERTENSION, MICROVASCULAR DISEASE AND DNA DAMAGE.
THE SPIKE PROTEIN CAUSES HUMANS TO “SELF-INJECT” THEMSELVES WITH ANG II. JUST AS IS DONE IN LAB MICE EXPERIMENTS
RAAS HYPERACTIVATION – ANG II “INFUSION”
https://wmcresearch.org/raas-hyperactivation-ang-ii-infusion/
The paper also proves viral entry (Spike Protein) causes this loss of ACE2.
Once the virus enters the host cell, it reduces ACE2 expression, leading to an increase in Ang II, which in turns binds to its receptor, Ang II receptor type 1, and influences the expression of various matrix metalloproteinases like MMP-1 and MMP-3, and inflammatory cytokines such as TNF-α, IL-6, IL-8, IL-1, and MCP1 through nuclear factor κB signaling [50,51].
This, as my article demonstrates, also induces many other pathologies and diseases of aging.
Furthermore, I proposed that these effects of the Spike Protein would induce systemic fibrosis, precisely parallel to that found in Radiation Fibrosis Syndrome.
SPIKE PROTEIN FIBROSIS SYNDROME
https://wmcresearch.org/spike-protein-fibrosis-syndrome/
The paper also shows this to be the case.
The microclots, composed of fibrin and other materials generated by vessel injury, persist for months following the subsidence of lung inflammation, indicating ongoing vascular injury. This could be partly aributed to oxygen deprivation due to fibrosis [68].
iii. These persistent symptoms are more likely to result in post-COVID-19 pulmonary fibrosis (PCPF). Interestingly, PCPF has been reported in approximately 44.9% of COVID-19 survivors. Patients with fibrosis frequently experience persistent symptoms such as dyspnea, cough, chest pain, fatigue, and myalgia [69]. Notably, the fibrosis pattern in COVID-19 differs markedly from that seen in occupational settings [70]. It is more uniform and less detectable by conventional imaging (i.e., X-ray), and indicates a distinct pathological mechanism. This uniform fibrosis could further exacerbate the lack of oxygen and contribute to vascular complications.
Persistent Vascular Complications in Long COVID: The Role of ACE2 Deactivation, Microclots, and Uniform Fibrosis
https://www.mdpi.com/2036-7449/16/4/42
All of the above points to my current major long term concern about reinfections or repeated exposures to the Spike Protein. Is this DIFFICULT TO DETECT (see above quote) microvascular fibrosis slowly, inexorably closing off the blood supply to our organs? Answers are needed immediately. As I mentioned earlier, we need to be looking for evidence of this in the healthy population, not just those with Long COVID. Please see my post from last week for further details.
I will investigate therapeutics to raise levels of ACE2. This may prove to be a valuable tool to ameliorate the loss of ACE2 that the Spike Protein induces.
Please enjoy your 4th of July holiday if you are in the States! And please enjoy your Thursday, wherever you are. Thank you, as always, for your support, readership and dialog.
This is great research. Many thanks for focussing on the heart of the problem, disruption of our delicate glycocalyx blood vessel linings, causing endothelial and vascular damage. Of note is also the high levels of platelets in lungs that also have ACE2 receptors that are activated by Spike protein, causing blood clots. What we need is universal access to Troponin and d-dimer blood tests to reveal the full extent of micro-clot damage and CRP tests for inflammation. We can’t fight this thing blind folded. Also we need Nattokinase and Bromaline to disperse the amyloid complexes. Thanks again for your research.
I do not know of a single person who is not vaxxed and who has had covid who has any of these problems.
The people with these problems are the fools who injected the Rat Juice