
Mitochondrial Carpet Bombing: The Endothelium is the Gateway through which the Spike Protein Devastates the Mitochondrial Landscape
Mitochondrial Carpet Bombing: The Endothelium is the Gateway through which the Spike Protein Devastates the Mitochondrial Landscape
This mitochondrial devastation leads to multi-organ destruction and failure and induces hyperaccelerated aging.

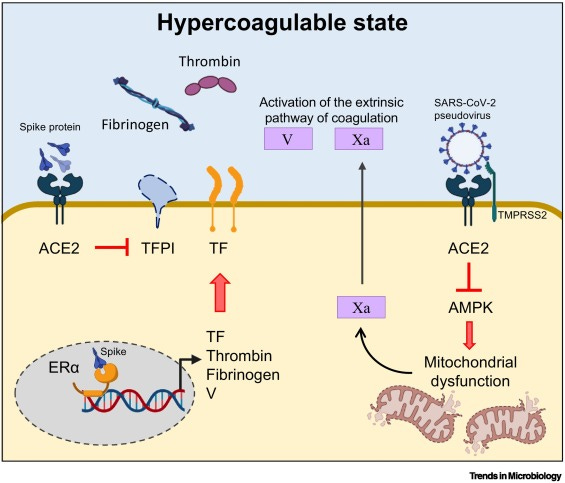
The spike protein of different SARS-CoV-2 variants inhibits tissue factor (TF) pathway inhibitor (TFPI), the primary regulator of the extrinsic pathway of blood coagulation. Concomitantly, the spike protein induces the transcriptional upregulation of TF, and increases the expression and secretion of coagulating factor V and thrombin. In addition, the spike protein strongly interacts with the human estrogen receptor α (ERα), possibly contributing to the upregulation of circulating factors involved in coagulation/fibrinolysis, such as coagulating factor V. Last, the direct binding of the spike protein to angiotensin-converting enzyme 2 (ACE2) induces the inactivation of adenosine monophosphate-activated protein kinase (AMPK), one of the key molecules regulating mitochondrial functions and biogenesis. Alterations in mitochondria lead to sustained metabolic dysregulation in endothelial cells, further altering the hypercoagulable state by upregulating coagulating factor Xa.
This is by far my most important work to date.
As readers of my Substack know, I have, from the beginning, hypothesized and demonstrated how the Spike Protein of SARS-CoV-2 induces what I have called Spike Protein Endothelial Disease. Now, with a great deal of further research I will demonstrate how this invasion of the Endothelium by the Spike Protein destroys the Mitochondria, further inducing organ damage. Given the evidence of how the Spike Protein destroys Mitochondria, this is extremely logical, as ACE2, the receptor of the Spike Protein, is ubiquitous.
The Spike Protein, in particular, induces alterations in mitochondrial metabolism IN MICROVASCULAR ENDOTHELIAL CELLS, which, as I have always maintained, is at the center of Spike Protein damage and pathology.
On top of that, intratracheal administration of the spike-expressing pseudovirus induced direct damage of lung endothelium by impairing mitochondrial function. Alterations in mitochondrial metabolism in human microvascular endothelial cells caused by the spike protein were accompanied by exuberant production of coagulating factor Xa, with obvious clinical implication for the hypercoagulable state observed in convalescent and severely ill COVID-19 patients. Data are available showing that the spike proteins of different SARS-CoV-2 variants, namely the Wuhan and Delta strains, induced transcriptional upregulation of tissue factor (TF), increased the expression and secretion of coagulating factor V, thrombin, and fibrinogen, and inhibited TF pathway inhibitor (TFPI), the primary regulator of the extrinsic pathway of blood coagulation, in human lung microvascular endothelial cells.
SARS-CoV-2 and the spike protein in endotheliopathy
https://www.cell.com/trends/microbiology/fulltext/S0966-842X(23)00189-0
This mitochondrial destruction is, of course, not limited to endothelial cells, the Sherman's March through the Mitochondria affects virtuall every organ and system.
Neuro-COVID
The Spike Protein, once it invades the microglia, it essentially "burns them out" by putting the cells into molecular overdrive, with disastrous consequences.

Effect of SARS-COV2 spike protein on NLRP3 and TNF- α expression in microglia. Results from the Immunofluorescence staining experiments showing representative images: Panel a: NLRP3 expression, Panel b: TNF- α expression that includes the following (i) Untreated control; (ii) Recombinant SARS-COV2 spike protein treated (iii) Quantitative histogram. Red stain in Panel A and B is Alexa Fluor 647 dye. Expression was quantitated using ImageJ software and statistical comparisons were done based on a comparison with the respective untreated control. A p value of < 0.05 is considered a statistically significant difference. Standard immunostaining protocols were followed. Data are representative images from 3 separate experiments done in triplicate.
Mitochondria play a central role in the COVID neuropathogenesis process by virtue of the fact that it is a major reactive oxygen species (ROS) producer, and a target of ROS. The proximity of mtDNA to the ROS-generating electron transport chain makes mtDNA susceptible to oxidative damage. Our data shows that treatment with both SARS-COV 2 spike protein and HI SARS-COV2 resulted in a significant increase in mitochondrial respiration. Figure 5 shows a significant increase in both basal and maximal OCR in response to both SARS-COV 2 recombinant spike protein and HI SARS-COV2. Our results suggest that SARS-COV2 increased mitochondrial respiration in microglia which could lead to bioenergetic dysfunction and production of ROS and increased oxidative stress. Host responses against SARSCOV2 depend on mitochondrial functions and mitochondrial DNA itself acts as a danger-associated molecular pattern (DAMP) and mitochondrial dysfunction drives a systemic immune response in COVID-19 pathogenesis. (Singh et al. 2020). Since mtDNA encodes vital components of the OXPHOS and protein synthesis machinery, oxidative damage-induced mtDNA mutations that impair either the assembly or the function of the respiratory chain will, in turn, further accumulation of ROS, which results in a vicious cycle leading to energy depletion in the cell and ultimately cell death. Our results show that SARS-COV2 increased mitochondrial respiration in microglia which could lead to bioenergetic dysfunction and production of ROS and increased oxidative stress.
Thus, our data suggests that SARS-COV2 induces a significant inflammatory response, increased oxidative stress, inflammasome activation and mitochondrial dysfunction in microglial cells, all of which contribute to COVID associated neuropathology. This study provides important mechanistic insights into SARS-COV2 induced mitochondrial dysfunction which underlies COVID-19 associated neuropathology.
Mitochondrial Dynamics in SARS-COV2 Spike Protein Treated Human Microglia: Implications for Neuro-COVID
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8487226/
The Heart

Proposed mechanisms through which S1 induced cardiac mitochondrial dysfunction which leads to cardiac injury in COVID-19 patients. Spike protein is the glycosylated protein that covers the surface of SARS-CoV-2 and binds to the host ACE2 receptor to mediate the viral cell entry. It is composed of S1 and S2 subunits that are responsible for ACE2 binding and membrane fusion, respectively. S1 possibly binds to ACE2 on the AC16 membrane, and is then internalized into the cytosol and localized in organelles, such as mitochondria, which induces the transient increase in fatty acids transport and uptake for biogenetics, Δψm, and permanent mCa2+, and disrupts Δψm later, finally impairing mitochondrial function and promoting ROS production. In turn, ROS further exacerbates mitochondrial function and mitochondrial fragmentation. Moreover, S1 also causes downregulation of TOM20; this effect might inhibit the pathways leading to mitochondrial biogenesis. ACE2, angiotensin converting enzyme 2; FAT, fatty acid translocase; PCT1/2, Carnitine palmitoyltransferase 1/2; MCD, Malonyl-CoA Decarboxylase; ACC, acetyl-CoA carboxylase; AMPK, AMP-activated protein kinase; mCa2+, mitochondrial Calcium, Δψm, mitochondrial membrane potential; ROS, reactive oxygen species.
Once again, we observe the Spike Protein "burning out" the cardiomyocytes. Interestingly, in the beginning, one may observe something "beneficial" as in the short term, this increased "power" improves certain functions. However, too much of a good thing sets in and the overloaded cells buckle, LITERALLY, under the pressure.
To the best of our knowledge, this is the first study to evaluate the effects of S1 on mitochondrial function in human cardiomyocytes. Although S1 improved mitochondrial function in AC16 cells in the short term, prolonged S1 treatment led to mitochondrial dysfunction due to the disruption of Δψm, mitochondrial Ca2+ overload, accumulation of ROS, and alteration of mitochondrial dynamics. These effects of subunit S1 of the S protein translate into irreversible cardiac remodeling.
Spike Protein Impairs Mitochondrial Function in Human Cardiomyocytes: Mechanisms Underlying Cardiac Injury in COVID-19
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10046940/
We now have a deeper understanding of how SPED, as I initially believed, is just the first stage in the development of a serious, debilitating condition. And, we now have an understanding of what that condition almost certainly is.
Essentially,this post is a synthesis of my work with SPED, uniting it with the work of the Salk Institute, which discovered the Spike Protein's damage of mitochondria in April of 2021.
The team then replicated this process in the lab, exposing healthy endothelial cells (which line arteries) to the spike protein. They showed that the spike protein damaged the cells by binding ACE2. This binding disrupted ACE2’s molecular signaling to mitochondria (organelles that generate energy for cells), causing the mitochondria to become damaged and fragmented.
The novel coronavirus’ spike protein plays additional key role in illness
https://www.salk.edu/news-release/the-novel-coronavirus-spike-protein-plays-additional-key-role-in-illness/
None of this would have been possible without your continued support and encouragement. I will continue to be BOLD and RELENTLESS. I will also be researching therapeutics to deal with this hellish dilemma, which, of course, never had to occur, nor should have.
Thank you
I have harped on about mitochondria for a long time now
The more people know about how these organelles are influenced and damaged
And what type of conditions/symptoms their dysfunction creates (depending on what tissue is damaged – the biodistribution)
The better.
Long Vax syndrome and Long Covid
Chronic fatigue syndrome
Chronic Lyme disease
Ciprofloxacin toxicity (Floxed)
Fibromyalgia
Gulf War Syndrome
POTS
Are likely all syndromes with mitochondrial dysfunction underpinning their onset
And these conditions do not always appear immediately
But creep up over years and come and go like waves and the tide.
It's a concerning subject
But pretending it isn't, isn't going to fix anything
The weaponry language in this article is apt. Enemy at the endothelium gate.