

Draining the Batteries: The Spike Protein Endothelium <> Mitochondria Feedback Loop and Sudden Cardiac Death
Mitochondrial dysfunction adversely impacts cardiac electrical functioning.

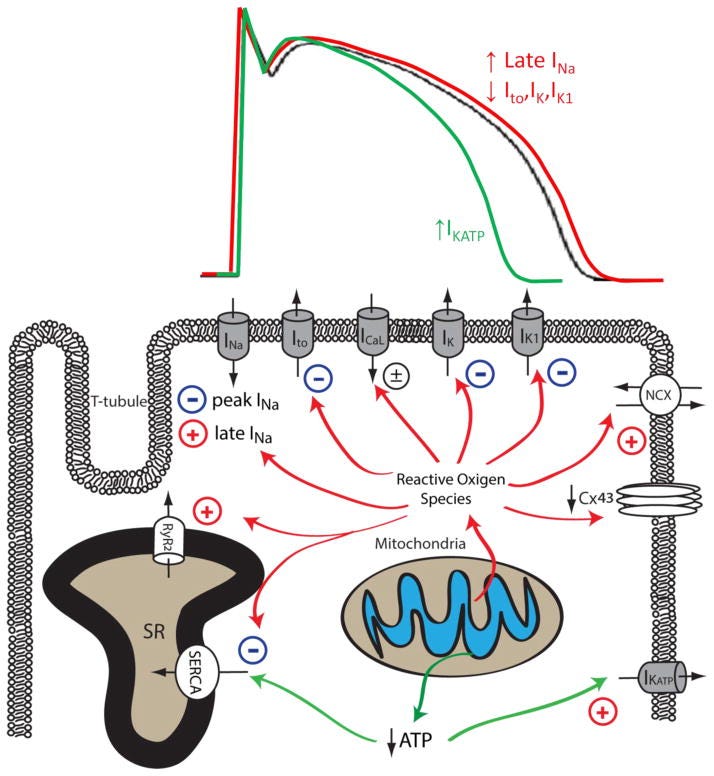
Mitochondrial dysfunction upon increased pathological or metabolic stress can lead to increased ROS production and reduced ATP synthesis. Increased cellular ROS can lead to the inhibition of peak INa, Ito, IK, IK1, SERCA and the downregulation of Cx43, whereas the activity of late INa, NCX and RyR are enhanced by increased oxidative stress. Reduced ATP production secondary to mitochondrial dysfunction can inhibit the activity of SERCA and increase the activity of sarcolemmal KATP channels. The impact of mitochondrial dysfunction on AP waveforms is illustrated on top: increased mitochondrial ROS production can prolong AP duration by increasing late INa and reducing repolarizing K+ currents (Red). Under conditions such as acute ischemia, mitochondrial ATP production is reduced, leading to opening of sarcolemmal KATP channel and AP shortening (Green). The Baseline AP waveform is shown in black.
As readers of this Substack know, yesterday I published a post discussing how the mutually destructive feedback loop of Spike Protein Endothelial damage inducing Mitochondrial damage which then further damages the Endothelium (etc.), explains why those with Long COVID suffer from exercise intolerance. I now believe it can also explain a portion of the observed Sudden Cardiac Deaths.
The more we understand the Spike Protein, the more ways we find it has to not only induxe long term fatal diseases, but also to cause Sudden Cardiac Death. There is, of course, Myocarditis. In addition, I have explained how the Spike Protein can cause Sudden Cardiac Death through denervation and brain stem damage.
The discovered feedback loop of mutual destruction between the Endothelium and the Mitochondria can also explain Sudden Cardiac Death (SCD). The mechanism is summarized here:
In summary, mitochondrial dysfunction is prevalent in arrhythmogenic cardiac diseases including cardiac hypertrophy, heart failure and myocardial ischemia. Reduced ATP synthesis and increased ROS production associated with mitochondrial dysfunction can lead to malfunction of various cellular mechanisms that are required to maintain normal electrical functioning and intracellular ionic homeostasis in cardiomyocytes. As summarized in Figure 2 (above) and Table 1, mitochondrial dysfunction can lead to reduced peak INa and downregulation of Cx43, resulting in abnormal conduction and increased propensity for re-entrant type cardiac arrhythmias. Increased cellular ROS also increases late INa and reduces repolarizing Kv currents, leading to impaired repolarization, prolonged APD, EADs, increased electrical heterogeneity in the myocardium, and increased arrhythmia susceptibility. Mitochondrial dysfunction also leads to disrupted intracellular Ca2+ homeostasis in cardiomyocytes, resulting in cytosolic Ca2+ overload and proarrhythmic DADs. Finally, mitochondrial dysfunction also causes the depolarization of ΔΨm and the opening of sarcoKATP channels, creating a current sink for the propagating depolarization wave, potentiating conduction block and arrhythmia.
Mitochondria and Arrhythmias
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4096785/
Not only does this explain the observed SCD, but also the Heart Failure observed in Long COVID and Spike related pathologies.
mROS (Mitochondrial ROS) drive both acute emergent events, such as electrical instability responsible for SCD, and those that mediate chronic HF remodeling, characterized by suppression or altered phosphorylation of metabolic, antioxidant, and ion transport protein networks.
Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure
https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.118.312708
Another factor of paramount importance is the fact that not only does the Spike Protein induce mitochondrial damage via the endothelium, it also increases cardiac ROS via its dysregulation of ANG-II and its additional mitochondrial injury via RAS activation.
Previously, we showed that a mouse model (ACE8/8) of cardiac renin–angiotensin system activation has a high rate of spontaneous ventricular tachycardia and sudden cardiac death secondary to a reduction in connexin43 level. Angiotensin-II activation increases reactive oxygen species (ROS) production, and ACE8/8 mice show increased cardiac ROS. We sought to determine the source of ROS and whether ROS played a role in the arrhythmogenesis.
In summary, we found that RAS activation resulted in mitochondrial injury, mitochondrial ROS production, reduction in Cx43, and increased arrhythmic risk. These changes were ameliorated by a mitochondria-targeted antioxidant but not agents targeted to other sources of cardiac oxidation or a general antioxidant. These results establish that ROS can be arrhythmogenic and elucidate a possible mechanism, whereby ROS can cause arrhythmia.
Mitochondria Oxidative Stress, Connexin43 Remodeling, and Sudden Arrhythmic Death
https://www.ahajournals.org/doi/10.1161/CIRCEP.112.976787
I believe this hypothesis may also explain why ATHLETES, PERFORMERS and PILOTS are experiencing a surge in SCD.
As far as a treatment/prevention measure, I plan to write tomorrow's Friday Hope on CoQ10.
For the past 2 years (after Pfizer x 2 and covid) my husband has motor issues in both his legs. No official diagnosis yet, even after so many tests. Probably demyelination or type of mnd. It is getting worse, slowly. Neurologist say impossible that vaccine could have been the cause. Ddimer and heart is ok. Thank you for this information and Friday hope. I now believe spike caused ROS and mitochondrial damage. If we know what we are fighting we might get to a solution. Living in a nightmare.
Methylene blue can be a big help with electron transport chain and ATPsynthase. There’s a lot of good info out there on MB. Might be of help with what you have found.