Does Persistent Spike Protein Drive an Obliterative Foam-Cell Arteriopathy (OFCA)?
We often think of foam cells as the hallmark of atherosclerosis in large arteries. But what if we've been looking in the wrong place?

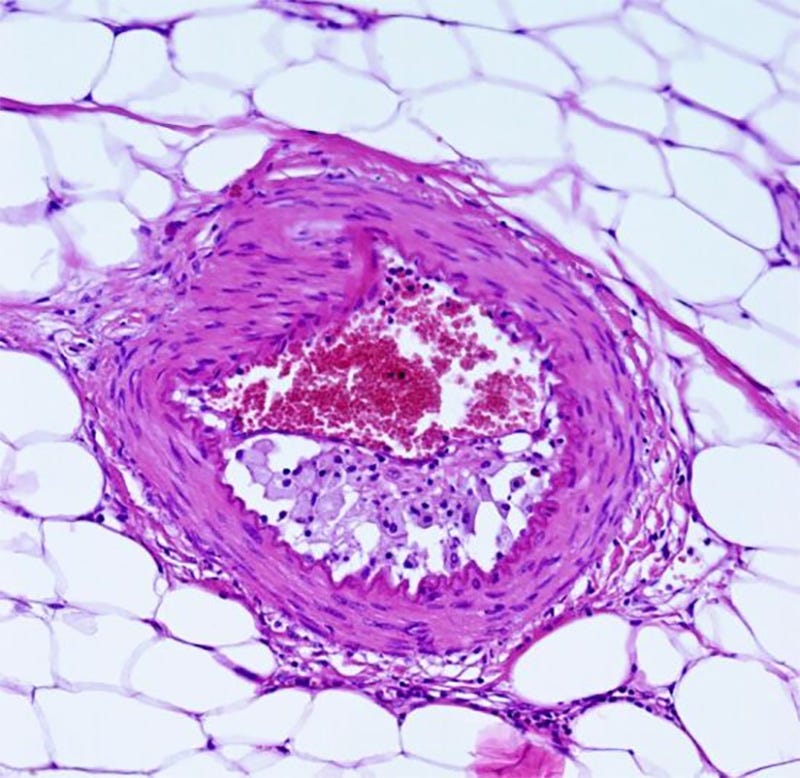
OFCA is characterized by the accumulation of lipid-laden macrophages in the intima causing narrowing. A thin adventitial cuff of fibrous tissue and mononuclear cell infiltration is present. HE staining, ×200. HE: Hematoxylin–Eosin; OFCA: Obliterative foam-cell arteriopathy
Recently, I came across a little-known vascular pathology called Obliterative Foam-Cell Arteriopathy (OFCA). The more I read, the more I wondered whether it might provide a framework for understanding persistent Spike Protein-induced vascular disease, especially since it is at the crossroads of so many Spike Protein-related pathologies.
OFCA is morphologically defined as a localized (single-organ) form of vasculopathy limited to small-sized or medium-sized arteries characterized by accumulation of lipid-laden macrophages in the intima causing narrowing or complete obstruction. OFCA is mainly a disease of the arterial intima. We propose to group a series of previously described entities, such as ionizing radiation arteriopathy, acute atherosis (foam-cell decidual arteriopathy), transplant arteriopathy of solid organ allografts, and intratumoral-associated foam-cell arteriopathy that constitute different manifestations of the same basic morphological process identified as OFCA. The basic mechanism of development of this arteriopathy is endothelial activation and dysfunction (local arterial endotheliopathy).
Obliterative foam-cell arteriopathy. A unifying concept embracing several entities previously described as radiation, decidual, transplant, and intratumoral-associated arteriopathy
https://pmc.ncbi.nlm.nih.gov/articles/PMC10863698/
Look at the last sentence in the above quote. Aren’t “endothelial activation and dysfunction” the quintessence of SPED (Spike Protein Endothelial Disease)?
The elements exist. The Spike activates the endothelium and causes endothelial dysfunction.
Although the use of a noninfectious pseudovirus is a limitation to this study, our data reveals that S protein alone can damage endothelium, manifested by impaired mitochondrial function and eNOS activity but increased glycolysis. It appears that S protein in ECs increases redox stress which may lead to AMPK deactivation, MDM2 upregulation, and ultimately ACE2 destabilization.4 Although these findings need to be confirmed with the SARS-CoV-2 virus in the future study, it seems paradoxical that ACE2 reduction by S protein would decrease the virus infectivity, thereby protecting endothelium. However, a dysregulated renin-angiotensin system due to ACE2 reduction may exacerbate endothelial dysfunction, leading to endotheliitis.
SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2
https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.121.318902
And the Spike Protein activates macrophages.
Results
We found TLR4 to be the most abundantly upregulated TLR in human lung tissue irrespective of the underlying pathology. Accordingly, bronchoalveolar lavage fluid cells from patients with severe COVID-19 showed an NF-κB-pathway dominated immune response, whereas they were mostly defined by type I interferon signalling in moderate COVID-19. Mechanistically, we found the Spike ectodomain, but not receptor binding domain monomer to induce TLR4-dependent inflammation in human macrophages. By using pharmacological inhibitors as well as CRISPR/Cas9 deleted macrophages, we identify SARS-CoV-2 to engage canonical TLR4-MyD88 signalling. Importantly, we demonstrate that TLR4 blockage prevents exaggerated inflammatory responses in human macrophages infected with different SARS-CoV-2 variants, including immune escape variants B.1.1.7.-E484K and B.1.1.529 (omicron).
Conclusion
Our study critically extends the current knowledge on TLR-mediated hyperinflammatory responses to SARS-CoV-2 in human macrophages, paving the way for novel approaches to tackle severe COVID-19.
SARS-CoV-2 activates the TLR4/MyD88 pathway in human macrophages: A possible correlation with strong pro-inflammatory responses in severe COVID-19
https://pmc.ncbi.nlm.nih.gov/articles/PMC10686889/
One finding especially caught my attention that really brought this hypothesis into focus. Investigators discovered that SARS-CoV-2 does not merely activate macrophages—it preferentially infects lipid-laden foam cells within human coronary plaques. Even more striking, infected foam cells produced a robust pro-atherogenic inflammatory response.
Here we show, in coronary autopsy specimens from patients with COVID-19, that infiltrating macrophages were infected by SARS-CoV-2. Lipid-laden macrophages (foam cells), a hallmark of atherosclerosis at all stages of the disease10, were more susceptible to SARS-CoV-2 infection than other macrophages, and this was dependent on the receptor neuropilin-1 (NRP-1). SARS-CoV-2 induced a strong pro-atherogenic inflammatory response in both macrophages and foam cells, which was largely recapitulated in an ex vivo SARS-CoV-2 infection of human vascular explants. This response may contribute to the ischemic cardiovascular complications in patients with COVID-19.
SARS-CoV-2 infection triggers pro-atherogenic inflammatory responses in human coronary vessels
https://www.nature.com/articles/s44161-023-00336-5
If foam cells themselves can harbor persistent viral material, they may function not simply as markers of vascular injury but as chronic inflammatory reservoirs capable of sustaining endothelial dysfunction long after the acute infection has resolved.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA generally becomes undetectable in upper airways after a few days or weeks postinfection. Here we used a model of viral infection in macaques to address whether SARS-CoV-2 persists in the body and which mechanisms regulate its persistence. Replication-competent virus was detected in bronchioalveolar lavage (BAL) macrophages beyond 6 months postinfection.
SARS-CoV-2 viral persistence in lung alveolar macrophages is controlled by IFN-γ and NK cells
https://www.nature.com/articles/s41590-023-01661-4
Putting all this together, here is what I propose may be occurring:
Persistent Spike Protein
↓
Chronic Endothelial Injury
↓
Oxidative Stress
↓
Oxidized LDL
↓
Foam Cell Formation
↓
IL-6 • ROS • TGF-β
↓
Further Endothelial Injury
↓
Progressive Luminal Narrowing
↓
Microvascular Rarefaction
↓
Chronic Tissue Ischemia
Of course, this remains a hypothesis, but one that is experimentally testable—and, if correct, could help explain the persistent vascular dysfunction observed in Long COVID and what may be occurring to some degree to all exposed to the Spike Protein. Thank you for keeping me going. I appreciate all of your support and am grateful for everyone in our community. Please have a blessed week.
One-Time Donation
https://buy.stripe.com/6oUbJ0auFaWkcLs2ZXeUU00
It is breathtaking to realize how much damage the spike protein construct can & is causing.
Dr Bhakdi said from the beginning that spike protein production with no off switch would cause huge damage to the endothelium, and that the
mRNA would create havoc in the genome. Wickedly subtle poisoning. But even a novice reader could spot the illogicality and overreach of the sales campaign.
Thank you for teasing out the detail of the project.